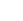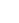專業科學儀器及設備制造商

DOI:10.1002/adma.202408906
全文速覽
在CO2加氫反應中,通常認為氧空位主要對CO2吸附和活化步驟起著重要作用。然而,根據氧空位的結構與電子特性,以及CO2轉化反應的連續性,氧空位對反應過程中一系列含氧中間體的作用可能長期被忽視和低估。本文以CO2光甲烷化為模型反應,利用富氧空位的Co3O4來研究氧空位與含氧中間體形成、轉化的關系。原位漫反射傅立葉變換紅外光譜(DRIFTS)分析和密度泛函理論(DFT)計算結果表明,甲酸鹽是反應過程的關鍵中間體,其C–O鍵斷裂是Co3O4表面甲烷化反應的限速步驟。而氧空位的存在可加速含甲酸鹽在內的各含氧中間體的C–O鍵斷裂及轉化,進而實現較高的CH4生成速率(1108.1 mmol g?1 h?1)和選擇性(93%)。這項研究為進一步認識CO2轉化反應中氧空位的多重作用提供了一定的見解,為后續設計和開發高性能CO2轉化催化劑提供了實驗支撐。
背景介紹
人為排放的溫室氣體CO2濃度不斷升高,造成了冰川融化,海平面上升等一系列環境問題,對自然環境構成了嚴重的威脅。光驅動的CO2加氫轉化為高附加值的燃料和化學品,被認為是轉化和利用溫室氣體CO2的有效策略。雖然氧空位被廣泛認為是氧化物基催化劑的活性位點,但關于其在CO2加氫過程中如何發揮作用的認知有限,迫切需要對其綜合作用進行深入的研究和揭示。為了解決這一問題,作者采用結構定制的Co3O4(富氧空位的介晶Co3O4,M-Co3O4)與商用Co3O4(C-Co3O4)作為對比,以CO2光甲烷化為模型反應,用于研究氧空位在含氧中間體形成和轉化過程中扮演的角色。
本文亮點
1. 本工作構建了具有特定晶面暴露特征的富氧空位Co3O4(M-Co3O4)。
2. 原位漫反射傅立葉變換紅外光譜(DRIFTS)分析和密度泛函理論(DFT)計算揭示了甲酸鹽(HCOO*)是關鍵中間體,其C–O鍵斷裂過程是Co3O4催化劑表面CO2光甲烷化反應的限速步驟。
3. M-Co3O4催化劑的氧空位加速了甲酸鹽及其它含氧中間體的C–O鍵斷裂,從而實現了優異的CH4生成速率(1108.1 mmol g–1 h–1)和高選擇性(93%)。
圖文解析
為了獲得特定晶面暴露的Co3O4催化劑,作者采用水熱和煅燒法構建了富含氧空位的介晶Co3O4(M-Co3O4,圖1)。M-Co3O4呈現出薄片穿插狀結構,并且在其矩形薄片中可以清晰觀察到大量的孔隙(圖1a–c,i–k)。通過透射電鏡分析得到M-Co3O4的晶格間距為0.28 nm,這與Co3O4的{220}晶面族十分吻合(圖1d)。從多孔矩形薄片的不同位置收集到的電子衍射斑點分布幾乎相同,表明M-Co3O4多孔薄片表現出介晶的性質(圖1e,f)。根據M-Co3O4多孔薄片的晶帶軸[111]以及測得的(![]() 02)和(2
02)和(2![]() 0)面,可推斷出M-Co3O4優先暴露(111)晶面。這種晶體結構配置可使M-Co3O4表面富集Co2+,并且更容易形成氧空位(圖1g,h)。
0)面,可推斷出M-Co3O4優先暴露(111)晶面。這種晶體結構配置可使M-Co3O4表面富集Co2+,并且更容易形成氧空位(圖1g,h)。

圖1.富氧空位M-Co3O4催化劑的結構表征
隨后,作者采用多種表征技術(如圖2所示),包括X射線光電子能譜(XPS)、電子順磁共振(EPR)、正電子湮滅譜(PAS)和X射線吸收精細結構譜(XAFS)等,來研究催化劑的化學狀態、電子結構、缺陷類型和配位環境,并藉此證實相比于商品Co3O4(C-Co3O4),結構定制的M-Co3O4含有大量的氧空位。
圖2. 鈷基催化劑的結構表征
圖3 鈷基催化劑的CO2加氫性能測試
為了深入揭示CO2加氫性能與催化劑表面氧空位之間的關系,作者在固定床反應器中使用CO2和H2的混氣(CO2/H2,1/4),對M-Co3O4和C-Co3O4催化劑進行了催化性能測試,如圖3所示。M-Co3O4催化劑展現出較高的CH4生成速率(1108.1 mmol g–1 h–1)及選擇性(93%)(圖3a)。這表明M-Co3O4在CO2加氫反應中活性優異,能夠有效地促進CH4的生成。相比之下,對照樣品也表現出較高的CH4生成速率(C-Co3O4為402.9 mmol g–1 h–1,CoO為217.1 mmol g–1 h–1,Co為282.2 mmol g–1 h–1),但均遠低于M-Co3O4。為了進一步確認CH4產物的C元素來源,作者以13CO2為C源進行同位素標記實驗,證實CH4是通過CO2加氫產生的(圖3b)。此外,M-Co3O4催化劑在5000 mL g–1 h–1的高空速條件下可持續運行30小時,CH4選擇性保持在96%以上,CO2轉化率高達54.04%,TON(以Co摩爾數為基)達66.17,這表明M-Co3O4具備優異的CO2加氫活性和良好的穩定性(圖3c)。
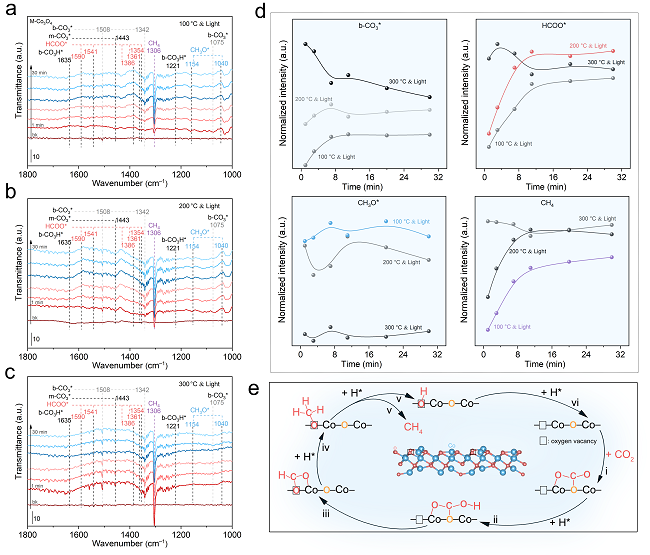
圖4. CO2加氫反應的DRIFTS機理研究
為了探索CO2加氫過程的反應機理和反應路徑,作者利用原位紅外光譜技術監測了CO2光加氫過程中Co3O4催化劑表面吸附物種和反應中間體的演化。當M-Co3O4和C-Co3O4催化劑暴露于CO2/H2(1/4)混氣時,可在其表面檢測到CO3H*、CO3*、HCOO*和CH3O*物種(圖4a–c)。圖4d展示了M-Co3O4催化劑表面各含氧中間體的演變趨勢。通過對上述各含氧中間體演變趨勢進行分析可知CO2光甲烷化的反應路徑為:CO2→CO3*/CO3H*→HCOO*→CH3O*→CH4。而HCOO*的演變趨勢很大程度上影響了最終產物CH4的生成,故推測HCOO*可能是CO2光甲烷化反應的關鍵中間體,其C–O鍵斷裂可能是限速步驟。
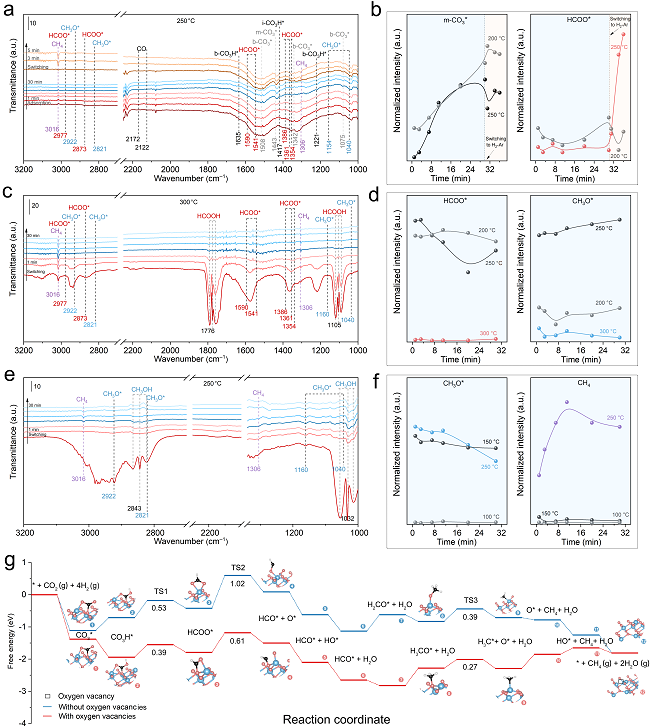
圖5. M-Co3O4催化劑表面碳酸鹽(CO3*)、甲酸鹽(HCOO*)和甲氧基(CH3O*)物種的轉化過程。
為了進一步闡明氧空位對各含氧中間體的作用,并證實HCOO*是CO2甲烷化反應的關鍵中間體,其C–O鍵的斷裂是反應限速步驟,作者開展了一系列原位DRIFTS實驗進行深入研究。具體來說,通過模擬反應中間體及反應氛圍(CO2 + H2/Ar、HCOOH + H2/Ar 和CH3OH + H2/Ar),并監測目標中間體的演化行為,以期證實HCOO*物種的緩慢轉化過程及氧空位在該過程中的獨特作用。
圖5a,b展示了M-Co3O4催化劑表面CO2吸附和轉化的情況。在溫度≤250°C時,b-CO3*物種立即形成并且保持基本恒定的濃度(圖5a,b);相比之下,m-CO3*物種的紅外峰面積(即濃度)隨著CO2暴露時間的增長而逐漸增大。將上述吸附CO2后的催化劑切換至H2/Ar氛圍后發現,當溫度為200°C時,b-CO3*和m-CO3*物種的轉化緩慢,而HCOO*及CH4均處于較低含量水平(圖5b);而當溫度達到250℃時,b-CO3*和m-CO3*物種含量逐漸衰減,與此同時HCOO*和CH4的紅外吸收逐漸增強(圖5a,b)。上述實驗表明b-CO3*和m-CO3*物種的臨界轉化溫度接近250℃,而在C-Co3O4表面也發現了類似的現象,盡管b-CO3*和m-CO3*物種的轉化以及CH4產物的形成要慢得多。
為了研究甲酸鹽(HCOO*)中間體的轉化行為并闡明氧空位的相關效應,作者將甲酸引入DRIFT系統,并監測其在H2/Ar氛圍下的相應變化(圖5c,d)。在≤250°C時,氣態HCOOH有微量消耗,HCOO*向CH3O*物種的轉化可以忽略不計,此時沒有檢測到CH4產物(圖5d);當溫度提升至300°C時,HCOO*和CH3O*物種立即處于低濃度水平,而CH4產物顯著增長(圖5c,d)。這些實驗結果表明,HCOO*物種的快速消耗和有效轉化發生在大約300°C。而在同等條件下測試氧空位較少的C-Co3O4時,HCOO*的消耗和CH4的生成速度較慢,這表明氧空位在HCOO*中間體的C–O鍵斷裂過程中發揮了促進作用。
針對CH3O*物種的轉化行為,作者將甲醇引入DRIFT系統,監測了其在H2/Ar氛圍下的轉化過程(圖5e,f)。在100℃和150℃時,盡管CH3OH和CH3O*物種的消耗速度不同,但并未觀察到CH4的生成(圖5f);相比之下,反應溫度為250℃時可立即檢測到氣相CH4產物(圖5e,f)。在C-Co3O4催化劑表面也得到了類似的實驗結果,盡管其CH4生成速度比M-Co3O4慢。這些分析結果表明,CH3O*物種所需的轉化溫度約為250°C或更低。
結合原位紅外實驗分析和DFT計算結果,我們描繪出氧空位作用下CO2甲烷化反應的完整過程(圖4e):i,游離CO2與M-Co3O4的晶格O結合形成CO3*;ii,活性H向CO3*轉移并生成CO3H*;iii,CO3H*進一步加氫裂解C–O鍵生成HCOO*,其中一個O原子填補氧空位;iv,HCOO*中連接Co位的C–O鍵斷裂,并逐漸氫化形成CH3O*,其O原子留在氧空位上;v,持續氫化導致CH3O*中C–O鍵斷裂,釋放CH4;vi,被O原子占據的氧空位通過H2的還原得到再生,從而完成催化循環。
總結展望
在這項研究中,作者采用富含氧空位的Co3O4(M-Co3O4)來深入探討氧空位在CO2光甲烷化過程中的具體作用。研究揭示了甲酸鹽(HCOO*)是反應過程的關鍵中間體,它的C–O鍵斷裂是CO2光甲烷化過程的限速步驟。M-Co3O4的氧空位加速各含氧中間體(包括CO3*/CO3H*、HCOO*和CH3O*)C–O鍵的斷裂,從而促進了其轉化,尤其是轉化較慢的甲酸鹽關鍵中間體,因此實現了優異的CH4生成速率。這項研究深化了對CO2轉化反應中氧空位關鍵作用的認識,為設計和優化高效CO2(光)加氫催化劑提供有價值的實驗支撐。
(TOC)
文章信息
Zhexing Lin, Zhengwei Yang, Jiajia Wang, Jun Wang, Huiting Huang, Jianyong Feng*, Huihui Yan, Minyue Zhao, Xinyi Liu, Wangxi Liu, Zhaosheng Li*, Zhigang Zou, Unlocking the Potential of Oxide-based Catalysts for CO2 Photo-hydrogenation: Oxygen Vacancies Promoted C–O Bond Cleavage in Key Intermediates, Adv. Mater. 2025, 2408906, https://doi.org/10.1002/adma.202408906
致謝
南京大學博士生林哲荇、博士生楊爭偉、河海大學副教授王家佳為該項成果共同第一作者。南京大學李朝升教授、馮建勇副教授為該論文通訊作者。該工作得到鄒志剛院士支持與指導。感謝王駿博士、黃輝庭博士、博士生閆會會、博士生趙敏躍、碩士生劉欣儀、博士生劉望喜在數據分析上提供的幫助。該工作受到國家自然科學基金(國家杰出青年科學基金)、國家重點研發計劃等項目支持,并得到固體微結構物理國家重點實驗室等平臺的大力支持。
本文使用的原位反應池由合肥原位科技有限公司研發,感謝老師支持和認可!